
The US Food and Drug Administration (FDA) has granted accelerated approval to zanubrutinib, a Bruton’s tyrosine kinase inhibitor, when combined with obinutuzumab in the treatment of patients with relapsed or refractory follicular lymphoma after two or more systemic lines of therapy.
The application for zanubrutinib, developed by BeiGene USA, Inc. under the name Brukinsa, was previously granted the Fast Track and Orphan Drug Designations from the FDA.
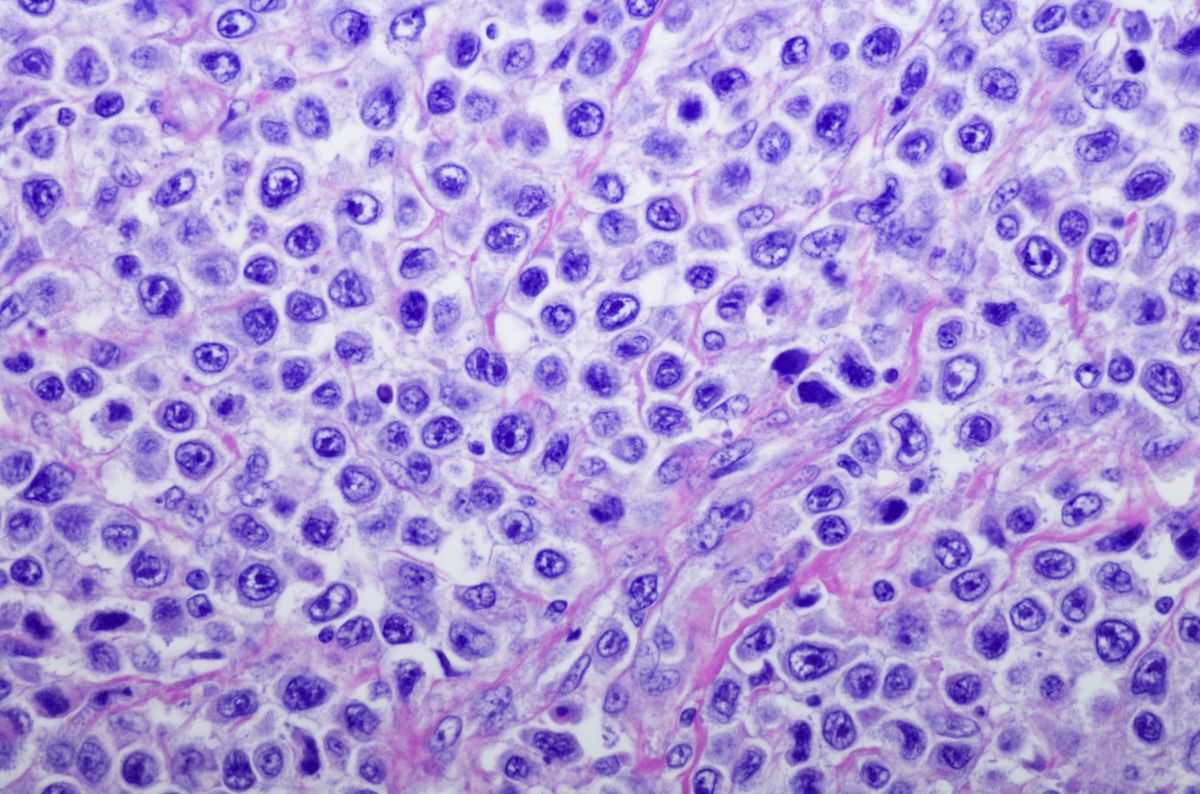
Approval for the therapy was supported by the ROSEWOOD study wherein 217 adult patients with a median of three lines of therapy (range, 2-11) received obinutuzumab alone or in combination with oral zanubrutinib 160 mg twice daily until disease progression or unacceptable toxicity.
The study estimated an overall response rate of 69% (95% CI, 61-76) in the zanubrutinib group versus 46% (95% CI, 34-58) in the obinutuzumab group. At a median follow-up of 19.0 months, the median duration of response was not reached (95% CI, 25.3-not estimable) in the zanubrutinib group versus 14.0 months (95% CI, 9.2-25.1) in the obinutuzumab group. The estimated 18-month duration of response in the zanubrutinib group was 69% (95% CI, 58-78).
Common adverse reactions to zanubrutinib observed across studies included decreased neutrophil and platelet counts, upper respiratory tract infection, hemorrhage, and musculoskeletal pain. Serious adverse reactions were reported in 35% of patients treated in the zanubrutinib group.




 © 2025 Mashup Media, LLC, a Formedics Property. All Rights Reserved.
© 2025 Mashup Media, LLC, a Formedics Property. All Rights Reserved.